What's on this Page?
- Missense3D-DB
- Structural features analysed for each variant
- How to use Missense3D-DB
- 3D structure selection
- The Missense3D algorithm
- Data availability
Missense3D-DB
Missense3D-DB is a database resource, which contains pre-computed atom-based calculations of the impact of amino acid substitution on protein structure obtained using the Missense3D algorithm. The current version of the database contains ~ 4 million missense variants from the following resources: Humsavar, ClinVar and gnomAD. Currently Missense3D-DB hosts variants prediction based on what we consider the best representative 3D coordinates for the query protein. Additional 3D coordinates representing the query protein in different conformational states or in complex with ligands or other proteins may be available. If you want to make predictions using different 3D coordinates please visit our variant prediction Missense3D software. Missense3D-DB is freely available to academic and commercial users.
Missense3D-DB is freely available to the scientific community and a paper [1] has been produced to describe this work.
1. Khanna T, Hanna G, Sternberg MJE, David A. 2021 Missense3D-DB web catalogue: an atom-based analysis and repository of 4M human protein-coding genetic variants. Published in Human Genetics, 140(5), 805-812. DOI: 10.1007/s00439-020-02246-z
Structural features analysed for each variant
The features that Missense3D checks for are listed below:
- Buried / exposed switch
- The substitution results in a change between buried and exposed state of the target variant residue. (RSA < 9% for buried and the difference between RSA has to be at least 5%.)
- Buried Gly replaced
- The substitution replaces a buried glycine.
- Buried H-bond breakage
- The substitution breaks all side-chain / side-chain H-bond(s) and/or side-chain / main-chain H-bond(s) formed by the wild type which was buried. The maximum H-bond N-O length is 3.9 angstrom.
- Buried Pro introduced
- The substitution introduces a buried proline.
- Buried charge introduced
- The substitution replaces a buried uncharged residue with a charged residue.
- Buried charge replaced
- The substitution replaces a buried charged residue with an uncharged residue.
- Buried charge switch
- The substitution switches the charge (+/-) of the buried residue.
- Buried hydrophilic introduced
- The substitution replaces a buried hydrophobic residue with a hydrophilic residue.
- Buried salt bridge breakage
- The substitution breaks a salt bridge formed by wild-type which was buried. The maximum N-O bond length is 5.0 angstrom.
- Cavity altered
- The substitution leads to an expansion or contraction of the cavity volume of > 70 angstrom^3. Cavity also refers to a pocket on the surface.
- Cis Pro replaced
- A cis proline in the wild type is replaced in the mutant.
- Clash
- The mutant structure has a MolProbity clash score > 30 and the increase in clash score is > 18 compared to the wild type.
- Disallowed phi/psi
- The mutant residue is in outlier region while the wild-type residue is in the favoured or allowed region.
- Disulphide breakage
- The substitution breaks a disulphide bond that was in the wild-type. The maximum S-S length for the bond is 3.3
- Gly in a bend
- The wild-type residue is glycine and is located in a bend curvature (reported 'S' in DSSP).
- Secondary structure altered
- A substitution results in a change in the DSSP secondary structure assignment at the variant position.
Hydrophobic residues are as follows: A, C, F, I, L, M, V and W; hydrophilic residues are as follows: D, E, H, K, N, Q and R, with the others being neutral (G, P, S, T and Y). D and E are treated as negatively charged and H, K and R as positively charged. We note that there are several variations to these definitions of residue properties.
How to use Missense3D-DB
1. The front page of Missense3D-DB allows the user to search Missense3D-DB database using either UniProt ID or Gene name.
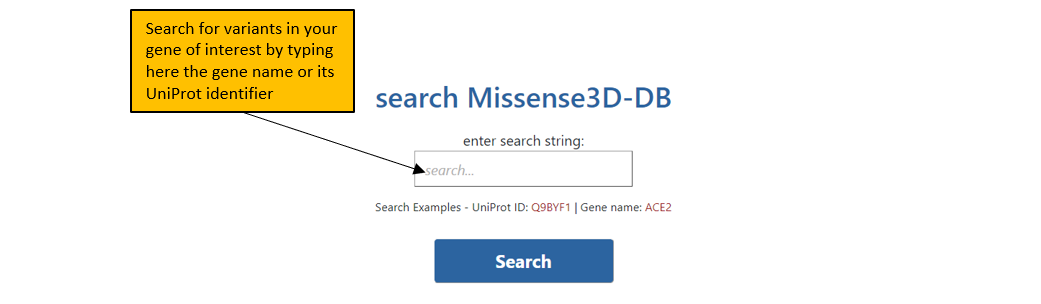
2. A valid input will direct you to the first result page which tabulates all the variants in Missense3D-DB for the input protein.

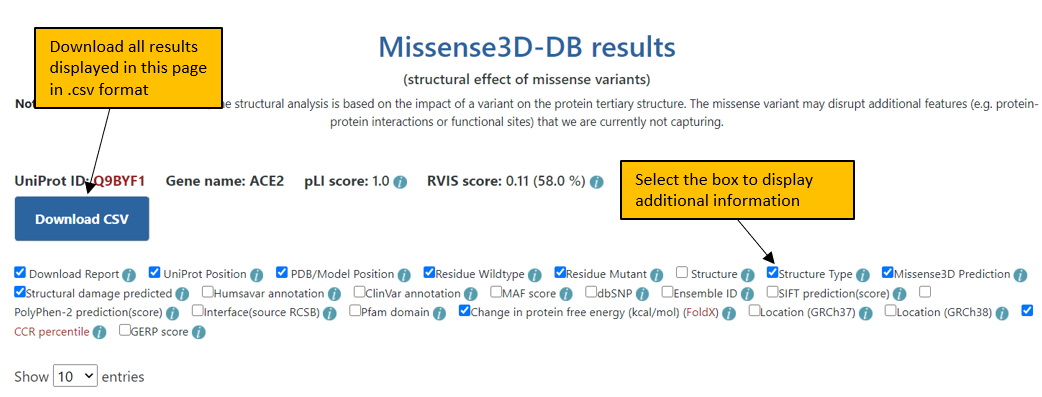
3. The final Results page shows the results for the input protein at a given UniProt position or all variants in Missense3D-DB. Additional columns of information can be displayed for the variants by selecting the appropriate box in the header. All the results displayed in this page can be downloaded in a csv format.
4. In the Results page, variants can also be ordered by clicking on a particular feature header. For example, to order variants according to the type of structure used for the analysis, click on “Structure type”.
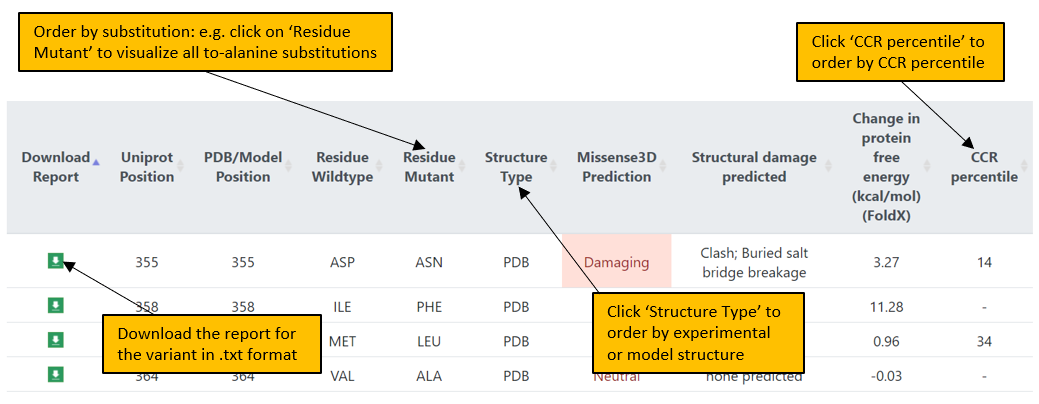
5. A page displaying the in-depth structural analysis can be accessed by clicking on the Missense3D prediction: Damaging or Neutral.

6. The structural analysis page. The 3D coordinates of the mutant structure can be downloaded from this page.

3D structure selection
We selected the best representative 3D coordinates based on the following criteria:
For a given UniProt ID and variant position:
- Group experimental PDB structures based on similarity to UniProt canonical sequence.
- Each group was further divided into 6 groups in order of preference:
- Group1: PDB structures with resolution ≤ 2.5Å
- Group2: PDB structures with resolution > 2.5Å and resolution ≤ 3.5Å
- Group3: PDB structures with resolution > 3.5Å
- Group4: NMR PDB structures
- Group5: EM PDB structures
- Group6: Other PDB structures
- The PDB structures with highest coverage covering the variant position were searched in order from the above groups. In case of a tie the highest resolution structure was chosen (e.g. given two structures, we would prefer a resolution of 1.8A to one of 2.5A).
- If the variant was not found in the experimental PDB structure then Phyre models were searched and the model with highest coverage was selected. All Phyre models are above 95% confidence with sequence identity > 30% and length of the modelled structure > 30 residues.
The Missense3D algorithm
Missense3D is prediction software, which allows the user to predict the structural impact of missense variants on protein structure. Missense3D provides binary prediction as structurally ‘Damaging’ or ‘Neutral’ based on 16 structural features like ‘breakage of salt bridge’, ‘cavity altered’ etc [2]. The salient feature of the software is that it produces comparable results with experimental structures and modelled structures.
2. Ittisoponpisan, S., Islam, S.A., Khanna, T., Alhuzimi, E., David, A. & Sternberg, M.J.E. (2019) Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 431, 2197-2212. DOI: 10.1016/j.jmb.2019.04.009
Data availability
The metadata for this project are available at https://doi.org/10.17605/OSF.IO/KBWRF
The wild type 3D structures (experimental and models) used for the analyses are available from the PhyreRisk website at http://phyrerisk.bc.ic.ac.uk/. The entire dataset of mutant structures generated for the analyses are available upon request on a case-by-case basis and subject to appropriate use. However, for each variant, the 3D coordinates of the mutant structure are available for download from the in-depth structural analysis page.