Documentation
Missense3D-TM
Missense3DTM predicts the structural impact of missense variants on transmembrane protein structure. Missense3DTM provides an overall binary prediction as structurally ‘Damaging’ or ‘Neutral’ based on 25 structural features like ‘breakage of a salt bridge’ and ‘cavity altered’. The salient feature of the software is that it works well on experimental structures and modelled structures.
Missense3D-TM is freely available to the scientific community and a paper [1] has been produced to describe this work.
1. Hanna G, Khanna T, Islam SA, David A, Sternberg MJE. 2023 Missense3D-TM: predicting the effect of missense variants in helical transmembrane protein regions using 3D protein structures. Awaiting Publication. DOI: 10.1007/s00439-020-02246-z
What's on this Page?
Structural features analysed for each variant
To view the features that standard Missense3D checks for, click
- Buried / exposed switch
- The substitution results in a change between buried and exposed state of the target variant residue. (RSA < 9% for buried and the difference between RSA has to be at least 5%.)
- Buried Gly replaced
- The substitution replaces a buried glycine.
- Buried H-bond breakage
- The substitution breaks all side-chain / side-chain H-bond(s) and/or side-chain / main-chain H-bond(s) formed by the wild type which was buried. The maximum H-bond N-O length is 3.9 angstrom.
- Buried Pro introduced
- The substitution introduces a buried proline.
- Buried charge introduced
- The substitution replaces a buried uncharged residue with a charged residue.
- Buried charge replaced
- The substitution replaces a buried charged residue with an uncharged residue.
- Buried charge switch
- The substitution switches the charge (+/-) of the buried residue.
- Buried hydrophilic introduced
- The substitution replaces a buried hydrophobic residue with a hydrophilic residue.
- Buried salt bridge breakage
- The substitution breaks a salt bridge formed by wild-type which was buried. The maximum N-O bond length is 5.0 angstrom.
- Cavity altered
- The substitution leads to an expansion or contraction of the cavity volume of > 70 angstrom^3. Cavity also refers to a pocket on the surface.
- Cis Pro replaced
- A cis proline in the wild type is replaced in the mutant.
- Clash
- The mutant structure has a MolProbity clash score > 30 and the increase in clash score is > 18 compared to the wild type.
- Disallowed phi/psi
- The mutant residue is in outlier region while the wild-type residue is in the favoured or allowed region.
- Disulphide breakage
- The substitution breaks a disulphide bond that was in the wild-type. The maximum S-S length for the bond is 3.3
- Gly in a bend
- The wild-type residue is glycine and is located in a bend curvature (reported 'S' in DSSP).
- Secondary structure altered
- A substitution results in a change in the DSSP secondary structure assignment at the variant position.
To see the additional features that Missense3D-TM specifically checks for, click
- TM head charge switch
- The substitution switches the charge (+/-) of the variant residue or replaces a charged residue with an uncharged residue within the lipid head region.
- TM charge replaced
- The substitution replaces a TM-exposed charged residue with an uncharged residue.
- TM charge introduced
- The substitution introduces a charged residue in the TM exposed region (solvent accessibility ≥ 0.09)
- TM hydrophilic introduced
- Replacement of a hydrophobic residue with a hydrophilic residue in the TM-exposed region (solvent accessibility ≥ 0.09).
- TM H-bond breakage
- The substitution breaks all side-chain / side-chain H-bond(s) and/or side-chain / main-chain H-bond(s) formed by the wild type residue which was in the TM exposed region (solvent accessibility ≥ 0.09). The maximum H-bond N-O length is 3.9 Å.
- TM salt bridge breakage
- The substitution breaks a salt bridge formed by the wild-type residue which was in the TM exposed region (solvent accessibility ≥ 0.09). The maximum N-O bond length is 5.0 Å.
- TM Pro introduced
- The substitution introduces an exposed proline residue within the TM region (solvent accessibility ≥ 0.09).
- TM Gly replaced
- The substitution replaces a TM-exposed glycine residue within the TM region (solvent accessibility ≥ 0.09).
- TM charge switch
- The substitution switches the charge (+/-) of the exposed residue within the TM region (solvent accessibility ≥ 0.09).
Hydrophobic residues are as follows: A, C, F, I, L, M, V and W; hydrophilic residues are as follows: D, E, H, K, N, Q and R, with the others being neutral (G, P, S, T and Y). D and E are treated as negatively charged and K and R as positively charged. We note that there are several variations to these definitions of residue properties.
How to use Missense3D-TM
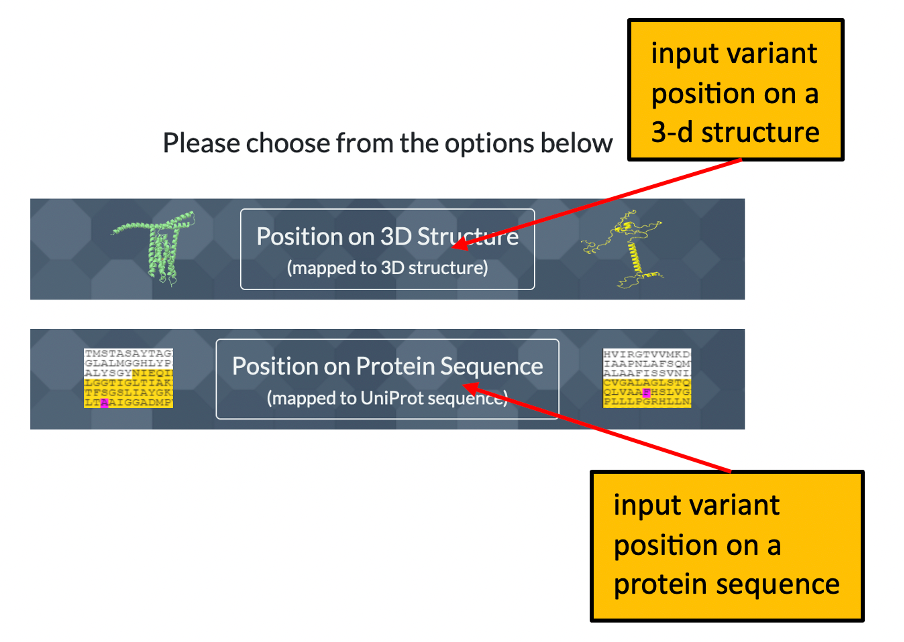
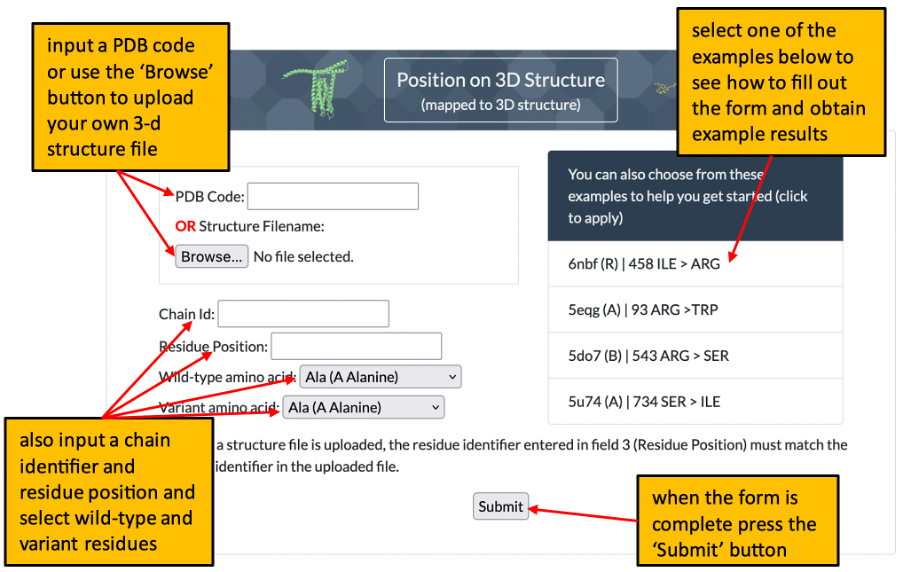
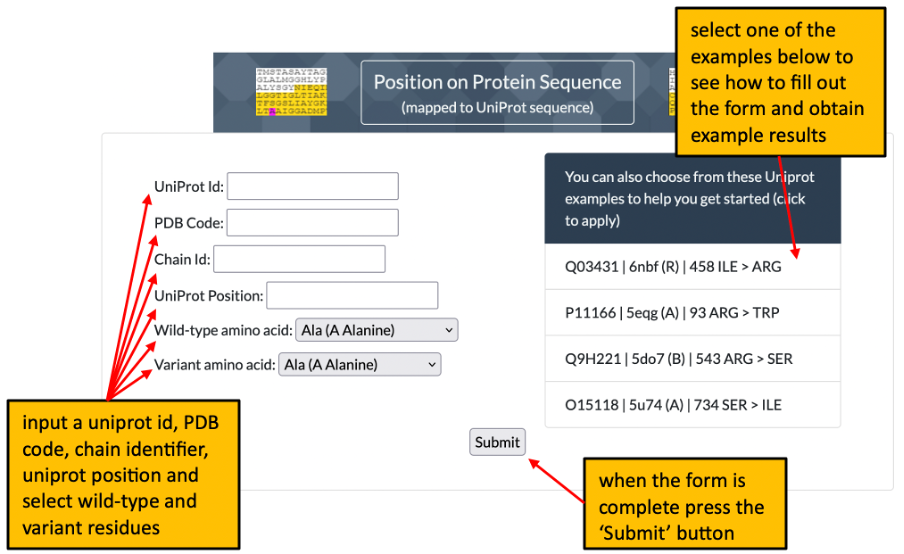
1. The front page of Missense3D-TM allows the user to enter details of a variant to analyse by using either its position on a structure or its position within a uniprot sequence. Please click here for an illustration.
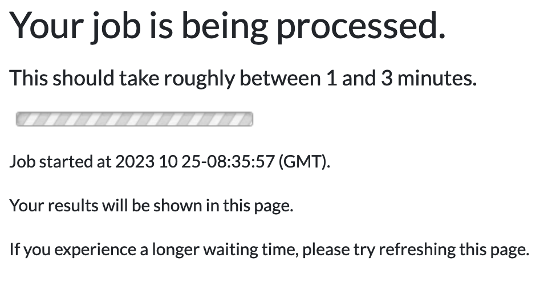
2. When you submit your request for variant analysis a waiting page will be displayed until the analysis completes. For details, please click here for an illustration.
3. Once processing of your request has completed you will be automatically directed to the summary results page which shows any pathogenic features identified and includes a link to the detailed results page for the current query. Please click here for further details of the summary page.

4. The detailed results page shows the results for the input variant at a given position in a UniProt sequence or at a given residue within a structure file. This page has a number of areas which contain a lot of information about the structure and the Missense3D-TM analysis. Please click here for help understanding the information in the detailed results page.

The Missense3D-TM algorithm
Missense3D-TM is prediction software which is based on the standard Missense3D algorithm but which has been extended to allow prediction of the structural impact of missense variants on transmembrane protein structures. Missense3D-TM provides binary prediction as structurally ‘Damaging’ or ‘Neutral’ based on the 16 structural features like ‘breakage of salt bridge’ present in standard Missense3D [2] but with 9 additional transmembrane-specific categories. The salient feature of this software is that it produces good results with both experimental structures and modelled structures.
2. Ittisoponpisan, S., Islam, S.A., Khanna, T., Alhuzimi, E., David, A. & Sternberg, M.J.E. (2019) Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 431, 2197-2212.
Data availability
All of the wild type and variant data and structures are available from the Missense3D-TM Dataset page